Highlights
Microsolvation of ions
Development of strategies for the study of solvation phenomena based on the formation of clusters.
In general, the global minimum structures are similar for both Li+Arn and Li+Krn. Modifications in the octahedral structure of the first solvation shell lead to a high-energy penalty. Conversely, the second solvation shell shows a panoply of minima with similar energies that are likely to be interconverted. Post-optimization at the MP2 level confirmed that, for n = 2 and 3, one has to include three-body terms in the potential to reproduce the low-energy structures.
Int. J. Quantum Chem. 119, e25860 (2019); doi: 10.1002/qua.25860
Aggregation of molecules
An evolutionary algorithm (EA) to discover the lowest-energy structures of Bzn clusters (for n=2-25), and the results are compared with previous global optimization studies where different potential functions were employed. Ab initio calculations carried out for the low-lying isomers of Bz3 and Bz4 clusters confirmed the global minimum structures for these sizes. The two lowest-energy Bz13 isomers show S6 and C3 symmetry, respectively, which is compatible with the experimental results available in the literature.
J. Comput. Chem. 36, 2291-2301 (2015); doi:10.1002/jcc.24201

Coronene is one of the basic polycyclic aromatic hydrocarbons (PAHs) used to test the reliabilty of a multidimensional potential energy surface (PES) and to assess its influence on the formation dynamics of PAH clusters with defined physical and chemical properties. Coronene clusters (Corn) are prototypes of PAHs. A large variety of low-energy structures were obtained for the Corn (n = 2−15) clusters ranging from columnar-type to two-stacked in a handshake association motifs. We found that a transition from a single-stack columnar regime to other more complex shapes occurs at n=6, whereas previous results based on a simpler coarse-grained potential pointed to a transition at n=8. Geometry reoptimizations were also performed at the DFT level for the most representative low-energy structures of Corn (n = 3−6), which confirmed the reliability of the present findings.
J. Phys. Chem. C 121, 14330-14338 (2017); doi: 10.1021/acs.jpcc.7b03691
Molecular modeling and simulations for environmental applications
Extensive use of pesticides causes serious environmental problems that require insightful knowledge from physics and chemistry sciences to be efficiently tackled. In particular, diffusion is a relevant property when evaluating the fluxes of pesticides in the groundwater. This work employed the best practices for the computation of diffusion coefficients from molecular dynamics simulations in the canonical ensemble. The investigation has been complemented with a study of the effect of the simulation time on the calculated diffusion coefficients. The methodology was successfully tested in the water benchmark system and, then, it was applied to obtain the diffusion coefficients of various pesticides (cymoxanil, imidacloprid, pirimicarb and glufosinate ion). The comparison of the calculated diffusion coefficients with recent experimental data reveals errors ranging from for the glufosinate ion and for cymoxanil. The results have been rationalized in terms of solute–solvent interactions.
J. Mol. Liq. 340, 117106 (2021); doi: 10.1016/j.molliq.2021.117106
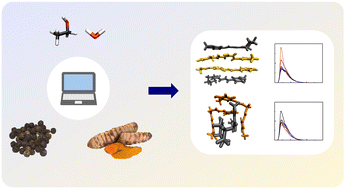
Molecular food chemistry
Molecular dynamics simulations on the aggregation patterns of curcumin and piperine in different polar media have shown a rapid formation of very stable dimers for both keto and enol tautomers in water. In agreement with a higher solubility, piperine molecules move closer and farther apart several times during the simulation, which indicates the formation of a less stable dimer in water. In contrast, both curcumin and piperine are soluble in ethanol and, thus, dimers can hardly be formed in this media. In comparison with a pure-water solvent, a 30 : 70 mixture of ethanol and water significantly reduces the probability of formation of most dimers of curcumin and piperine molecules. The simulations show that larger clusters may be complex structures, but the formation of stacks (in the case of piperine and enol tautomer of curcumin) and cages (when the keto tautomer of curcumin is involved) are not rare. Furthermore, it is shown that each single molecule presents a certain degree of mobility in the cluster, especially on the surface, but without leading to dissociation.
Phys. Chem. Chem. Phys. 25 (2023) 19899–19910; doi: 10.1039/d3cp00096f
Software development
We developed and implemented a superimposing algorithm to identify enantiomeric structures. The methodology was tested by applying it to several organic molecules and water clusters that were subjected to geometry optimization.
J. Chem. Inf. Model. 50, 2129-2140 (2010); doi: 10.1021/ci100219f
We developed a software package based on a genetic algorithm that fits an analytic function to a given set of data points. The code, called GAFit, was also interfaced with the CHARMM and MOPAC programs in order to facilitate force field parameterizations and fittings of specific reaction parameters (SRP) for semiempirical Hamiltonians. The present tool may be applied to a wide range of fitting problems, though it has been especially designed to significantly reduce the hard work involved in the development of potential energy surfaces for complex systems.
Comput. Phys. Commun. 217, 89-98 (2017); doi:10.1016/j.cpc.2017.02.008
We proposed an evolutionary algorithm (EA) for global optimization of binary atomic clusters. The method was applied to binary Lennard-Jones (BLJ) clusters as a testing ground, while results for mixed argon-krypton clusters were also reported. We show that the method is effective in discovering all global minima of BLJ clusters for nuclearities up to N=50 and different values of the atoms size ratio. Moreover, one new global minimum has been discovered for N=38 and size ratio 1.05.
Chem. Phys. Lett. 485, 211-216 (2010); doi: 10.1016/j.cplett.2009.11.059
