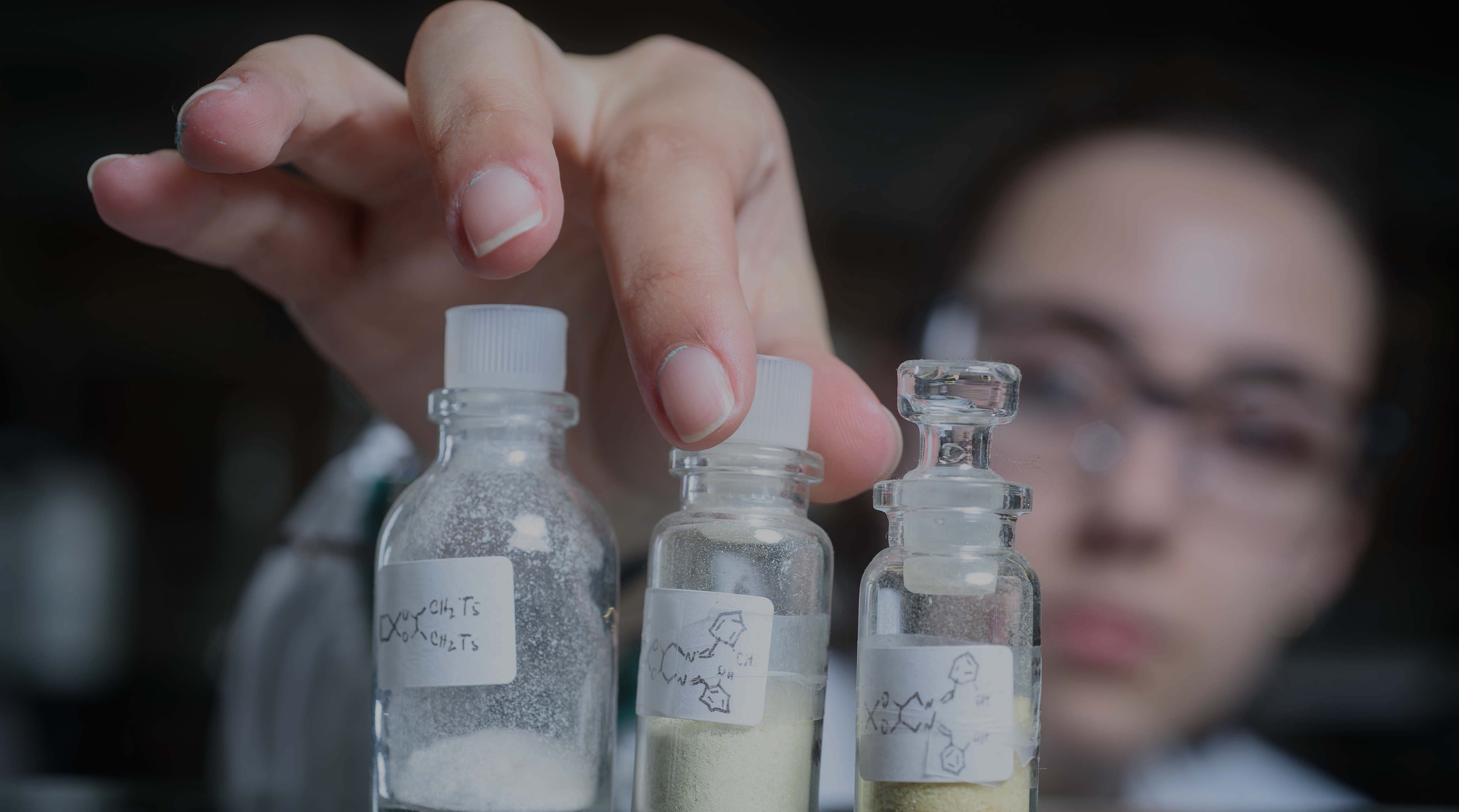
Pharmaceutical nanococrystal synthesis: a novel grinding approach

Abstract
Pharmaceutical nanococrystals are solid forms with potential to increment the oral bioavailability of drugs belonging to class II of the biopharmaceutical classification system through the conjunction of the advantages of cocrystal formation and particle size reduction. Herein, results regarding the investigation of in situ synthesis of these nanoparticles are reported, using (2 : 1) (S)-naproxen–nicotinamide as a cocrystal model, through grinding assisted by minimal amounts of solutions of non-ionic surfactants or by PEG 6000, as stabilizers. Additionally, studies regarding a top-down wet milling method were also carried out. Results identify the nature and concentration of the stabilizer and, for nanodispersions, the choice of dispersion medium, as major challenges in the synthesis of nanococrystals. Solid forms were characterized by attenuated total reflectance-infrared spectroscopy, differential scanning calorimetry and X-ray powder diffraction. Particle size was determined by scanning electron microscopy and laser diffraction, proving the successful obtention of a nanopowder (surfactant-assisted grinding, SAG) and a nanodispersion (wet milling) stabilized by a mixture of Span® 85 and Tween® 85. The one-step synthesis of nanococrystals through SAG proved to be a greener and efficient way to obtain these nanoparticles, paving the way for an important evolution in cocrystal nanosizing.

Abstract
In this work, solid state characterization and the investigation of the thermal behavior of cis-1,3 and trans-1,3 cyclohexanediol isomers were carried out. A plastic crystal phase could be identified only for the trans isomer, which vitrifies on cooling, while for the cis isomer two monotropically related ordered crystal phases have been found. These results allow gaining of comprehensive knowledge of the six cyclohexanediol isomers' plastic crystal forming ability; plastic crystal phases were observed for three of the six cyclohexanediol derivatives, coinciding with the axial–equatorial isomers cis-1,2, trans-1,3 and cis-1,4-cyclohexanediols. The molecular structure of all the possible conformations of the isolated molecules of the six cyclohexanediol isomers was optimized at the MP2/aug-cc-pVDZ level of theory. The axial–equatorial isomers have several conformers with relevant abundance, which can have greater entropy of mixing, which may have a role in stabilizing the disordered plastic phases. For trans-1,4-cyclohexanediol, the conformer population breakdown is 66.4% bi-equatorial and 33.6% bi-axial. The bi-axial conformations of trans-1,4-cyclohexanediol are preserved in the solid forms. The Hirshfeld surface analysis of the parent anisotropic phase was confirmed as a valuable tool to assess the plastic crystal forming ability in this family of compounds, and the asphericity parameter to be a good indicator. The fingerprint plots highlight frequent intermolecular H–H close contacts for cis-1,3-cyclohexanediol, also found in other non-plastic crystal former cyclohexanediols.
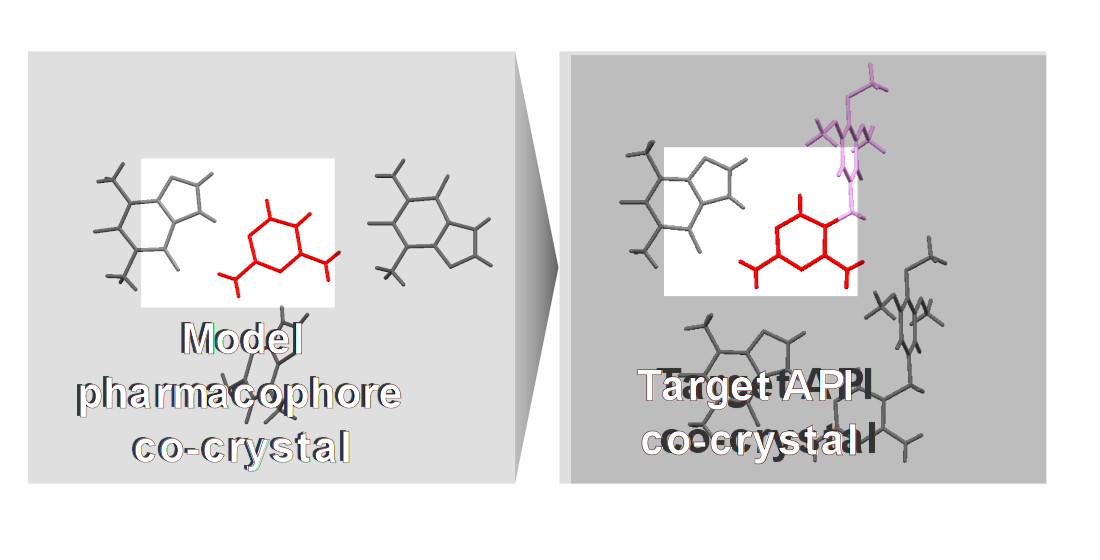
Dihydrofolate Reductase Inhibitors: The Pharmacophore as a Guide for Co-Crystal Screening
Abstract
In this work, co-crystal screening was carried out for two important dihydrofolate reductase (DHFR) inhibitors, trimethoprim (TMP) and pyrimethamine (PMA), and for 2,4-diaminopyrimidine (DAP), which is the pharmacophore of these active pharmaceutical ingredients (API). The isomeric pyridinecarboxamides and two xanthines, theophylline (THEO) and caffeine (CAF), were used as co-formers in the same experimental conditions, in order to evaluate the potential for the pharmacophore to be used as a guide in the screening process. In silico co-crystal screening was carried out using BIOVIA COSMOquick and experimental screening was performed by mechanochemistry and supported by (solid + liquid) binary phase diagrams, infrared spectroscopy (FTIR) and X-ray powder diffraction (XRPD). The in silico prediction of low propensities for DAP, TMP and PMA to co-crystallize with pyridinecarboxamides was confirmed: a successful outcome was only observed for DAP + nicotinamide. Successful synthesis of multicomponent solid forms was achieved for all three target molecules with theophylline, with DAP co-crystals revealing a greater variety of stoichiometries. The crystalline structures of a (1:2) TMP:THEO co-crystal and of a (1:2:1) DAP:THEO:ethyl acetate solvate were solved. This work demonstrated the possible use of the pharmacophore of DHFR inhibitors as a guide for co-crystal screening, recognizing some similar trends in the outcome of association in the solid state and in the molecular aggregation in the co-crystals, characterized by the same supramolecular synthons.
Pharmaceutical nanococrystal synthesis: a novel grinding approcah

Abstract
Pharmaceutical nanococrystals are solid forms with potential to increment the oral bioavailability of drugs belonging to Class II of the Biopharmaceutical Classification System, through the conjunction of the advantages of cocrystal formation and particle size reduction. Herein, results regarding the investigation of in situ synthesis of these nanoparticles are reported, carried out using (2:1) (S)-naproxen-nicotinamide as a cocrystal model,1 through grinding, assisted with minimal amounts of solutions of non-ionic surfactants or with PEG 6000, as stabilizers. Additionally, studies regarding a top down wet milling method were also carried out. Results identify stabilizer nature, stabilizer concentration and, for nanosdispersions, the dispersion media choice, as major challenges for the synthesis of nanococrystals. Solid forms were characterized by attenuated total reflectance-infrared spectroscopy, differential scanning calorimetry and X-ray powder diffraction, and particle size was determined by scanning electron microscopy and laser diffraction, proving the successful obtention of a nanopowder (surfactant assisted grinding, SAG) and a nanodispersion (wet milling), stabilized by a mixture of Span® 85 and Tween® 85. The one-stepped synthesis of nanococrystals through SAG proved to be a greener and efficient way to attain these nanoparticles, paving the way for an important evolution in cocrystal nanosizing.
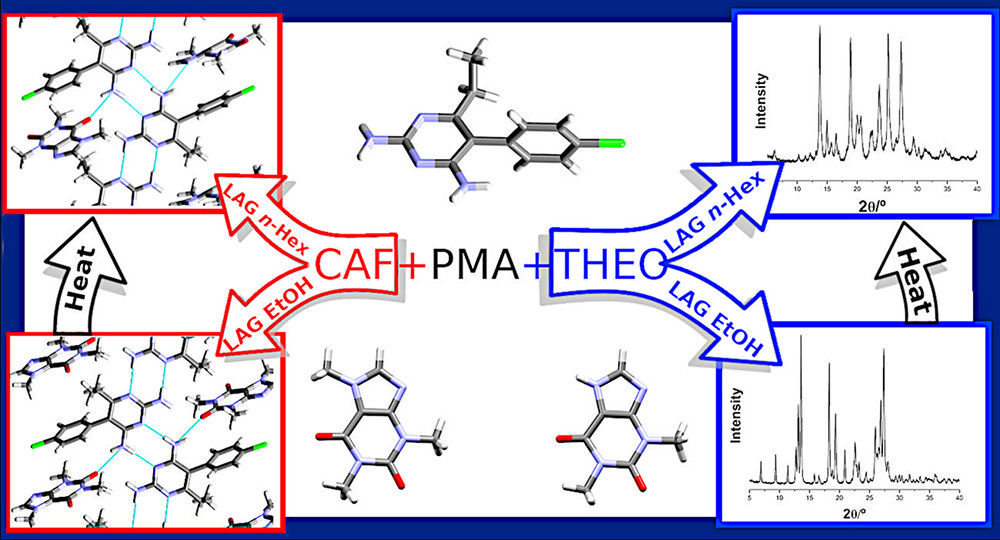
Polymorphic Cocrystals of the Antimalarial Drug Pyrimethamine: Two Case Studies
Abstract
Polymorphism is known to have significant implications in the pharmaceutical industry. However, there are relatively few studies of polymorphic pharmaceutical cocrystals, and contributions to the systematic assessment, structural, and thermodynamic characterization of cocrystal polymorphs are of great relevance. This work reports cocrystal formation between pyrimethamine, a multifunctional API with a 2,4-diaminopyrimidine scaffold, common to many dihydrofolate reductase inhibitors, and two structurally related coformers, caffeine and theophylline, differing in their hydrogen-bonding donor ability. Two (1:1) enantiotropic cocrystal polymorphs were identified for both pyrimethamine:caffeine and pyrimethamine:theophylline. The contribution of differential scanning calorimetry to guide the cocrystal polymorphs discovery should be emphasized. The different hydrogen-bonding abilities of the coformers have consequences for the crystalline structures obtained. The crystalline structure of pyrimethamine:theophylline polymorph II, previously solved by Delori et al., is determined by the NPMA···H–NTHEO bond. Both pyrimethamine:caffeine polymorphs, whose crystalline structures were solved in this work, although differing in packing and synthon, share a common 2,4-diaminopyrimidine hydrogen-bonded chain pattern which has quite close similarity with pyrimethamine polymorph I. A similar infrared pattern in the N–H stretching region is observed for these three solid forms, and also for pyrimethamine:theophylline, polymorph I, which may anticipate a similar arrangement in this polymorph.
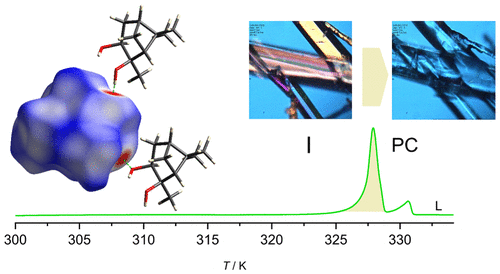
Ordered and Plastic Crystals in the Complex Polymorphism of Pinanediol
Abstract
Plastic crystals, frequently observed for compounds with globular molecules, are mesophases characterized by orientational disorder maintaining positional order. The present work focuses on finding correlations between the plastic crystal formation ability of pinanediol, its molecular shape, and the intermolecular interactions existing in solid phases. Pinanediol, a bicyclical monoterpenoid and vicinal diol, is a globular molecule capable of forming intermolecular hydrogen bonds. In the present work, the structure and thermal behavior of this compound in the solid state is studied for the first time. Despite its strong intermolecular interactions in condensed phases, it was found to be a plastic crystal former showing complex polymorphism. Three ordered phases were identified by differential scanning calorimetry, polarized light thermal microscopy, and X-ray diffraction with variable temperature. The molecular dynamics in different polymorphic forms was studied by broadband dielectric spectroscopy (BDS). The structure of the most stable enantiopure ordered polymorph of pinanediol was resolved by single-crystal X-ray diffraction, using crystals of both enantiomers. Its intermolecular close contacts were studied by Hirshfeld surface analysis, which was used to determine shape parameters correlated to plastic crystal formation ability. It was concluded that the globular shape of pinanediol allows the formation of the plastic phase that is not inhibited by the presence of intermolecular hydrogen bonding.
Lamotrigine: Design and synthesis of new multicomponent solid forms
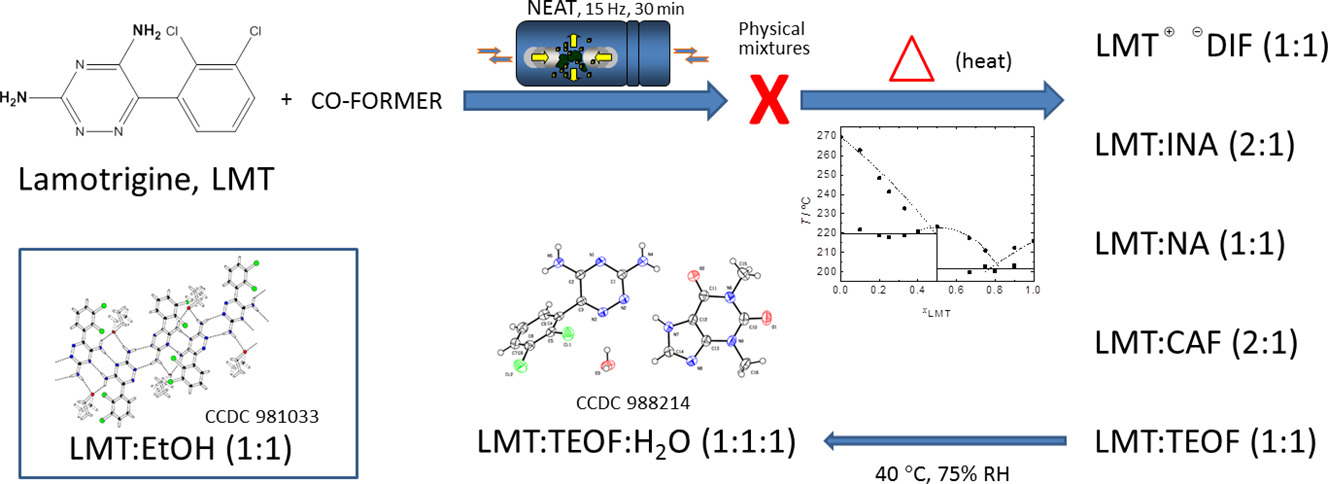
Abstract
In this work, a crystal engineering and thermodynamic based approach has been used aiming at contributing to a deeper knowledge of lamotrigine multicomponent solid forms. Two types of co-molecules have been chosen that can give rise to co-crystals with lamotrigine through different supramolecular heterosynthons: the xanthines, theophylline and caffeine, and the three isomeric pyridinecarboxamides. Association with diflunisal, which may result in a salt, was also investigated. Mechanochemistry, differential scanning calorimetry, thermogravimetry, X-ray powder and single crystal diffraction, infrared spectroscopy were the methods used. For all the systems, exploratory neat mechanochemistry experiments, carried out on lamotrigine + co-molecule binary mixtures of different compositions, were not successful in promoting association. From differential scanning calorimetry data and the binary solid-liquid phase diagrams, co-crystals/salts were identified as well as their respective stoichiometry, and a methodology of synthesis was established. For pyridinecarboxamides, molecular recognition is dependent on the position of the amide group in the pyridine ring: co-crystallization did not occur with picolinamide co-former. Both xanthines form co-crystals with lamotrigine, (1:1) with theophylline and (2:1) lamotrigine:caffeine. Additionally, the crystalline structure of a lamotrigine:theophylline 1:1 monohydrate was solved. The (1:1) lamotrigine:theophylline co-crystal converts to this monohydrate in accelerated stability tests. A (1:1) lamotrigine:diflunisal salt was identified, which proved to be stable in accelerated stability assays.